
Comunicări directe către profesioniștii din domeniul sănătății. TRISENOX (trioxid de arsen) - Risc de erori de medicație datorită introducerii concentrației de 2 mg/ml
O nouă concentrație: va fi introdus flaconul de 2 mg/ml pentru a înlocui fiola de sticlă autorizată de TRISENOX 1 mg/ml existentă.
Stimate profesionist din domeniul sănătății,
De comun acord cu Agenția Europeană pentru Medicamente (EMA) și Agenția Națională a Medicamentului și a Dispozitivelor Medicale din România ( ANMDMR), compania Teva Pharmaceuticals S.R.L. dorește să vă informeze cu privire la următoarele:
Rezumat
- Există riscul unor erori de dozare datorită introducerii unei noi forme de prezentare pentru TRISENOX (trioxid de arsen) care conține o concentrație dublă a substanței active.
- O fiolă de unică folosință de 10 ml care conține 1 mg/ml substanță activă (10 mg trioxid de arsen), va fi înlocuită
- Cu un flacon de 6 ml de unică folosință care conține 2 mg/ml substanță activă (12 mg trioxid de arsen).
- Cele două concentrații diferite pot coexista temporar pe piață și acest lucru poate duce la confuzii între cele două produse și la erori de medicație, fie cu risc de supradozaj cu potențial rezultat letal, fie cu risc de subdozaj cu potențială lipsă de eficacitate.
- Verificaţi întotdeauna cu atenţie concentraţia de TRISENOX atunci când calculaţi volumul care trebuie extras pentru diluare şi perfuzie, pentru a vă asigura că pacientului i se administrează doza corectă de trioxid de arsen.
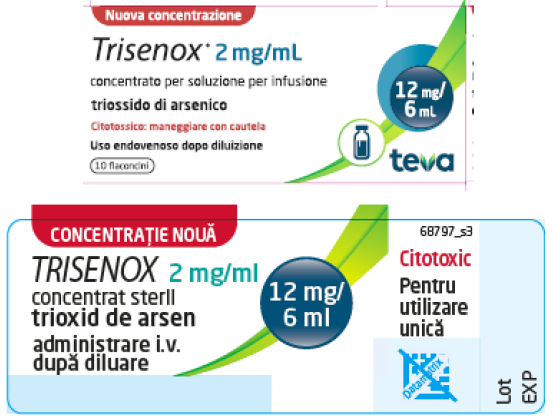
- Recunoașterea concentrației de medicament va fi facilitată de două pachete diferite, care sunt prezentate în tabelul următor:
| TRISENOX 1 mg/ml concentrat pentru soluție perfuzabilă, disponibil în prezent | TRISENOX 2 mg/ml, concentrat pentru soluție perfuzabilă, disponibil în viitor | |
| Concentrație | 1 mg/ml | 2 mg/ml |
| Unitate de ambalare | Fiolă de 10 ml | Flacon de 6 ml |
| Trioxid de arsen per recipient | 10 mg | 12 mg |
| Eticheta recipientului primar |  |
 |
| Partea frontală a cutiei de carton |  |
 |
| Reconstituire | Ambele pot fi diluate cu 100 până la 250 ml soluție injectabilă de glucoză 50 mg/ml (5%) sau soluție injectabilă de clorură de sodiu 9 mg/ml (0,9%) | |
Observație:
Produsul Trisenox 2 mg/ml va fi disponibil inițial având ambalajul inscripționat în limba italiană, urmând ca ulterior, produsul Trisenox 2 mg/ml să fie disponibil cu ambalajul inscripționat în limba română.
Informații suplimentare referitoare la problema de siguranță
TRISENOX este indicat pentru inducerea remisiunii și consolidare la pacienții adulți care prezintă:
- leucemie acută promielocitară (LAP) cu risc scăzut până la intermediar, nou diagnosticată (număr de leucocite ≤10 x 103 /μl) în asociere cu acidul all-trans-retinoic (AATR)
- leucemie acută promielocitară (LAP) recurentă/refractară (tratamentul anterior trebuie să fi inclus un retinoid și chimioterapie) caracterizată prin prezența translocației t(15;17) și / sau prezența genei leucemiei promielocitare/receptorului-alfa al acidului retinoic (LPM/RAR-alfa).
Erorile de dozare a medicamentului care rezultă din modificarea concentrației medicamentului disponibil pe piață și confuzia celor două forme de prezentare ale produsului pot duce la următoarele consecințe:
- Risc de supradozaj: potențarea unuia sau a tuturor riscurilor cunoscute asociate cu TRISENOX, care poate duce la un rezultat potențial letal în următoarele evenimente:
- Hemoragii abundente datorate trombocitopeniei;
- Infecții severe, sepsis și șoc septic din cauza leucopeniei severe;
- Stop cardiac brusc din cauza prelungirii intervalului QTc;
- Sindromul de diferențiere a leucemiei acute promielocitare (LAP );
- Sângerări intracraniene sau infarct miocardic ischemic din cauza hiperleucocitozei;
- Potențială leziune renală acută sau insuficiență renală din cauza toxicității renale crescute;
- Insuficiență hepatică potențială din cauza valorilor crescute ale transaminazelor hepatice, bilirubinei și gama-glutamiltransferazei din sânge.
Vă rugăm să citiți secțiunea 4.9 „Supradozaj” din rezumatul caracteristicilor produsului (RCP) pentru a afla cum să gestionați supradozajul.
- Risc de subdozaj: răspuns insuficient la tratament care duce la posibilitatea rezistenței tumorii la chimioterapie cu răspuns clinic redus.
Apel la raportarea de reacţii adverse
Este important să raportaţi orice reacţii adverse suspectate, asociate cu administrarea medicamentului TRISENOX către Agenţia Naţională a Medicamentului şi a Dispozitivelor Medicale din România, în conformitate cu sistemul naţional de raportare spontană, utilizând sistemul electronic de raportare/formularul de raportare disponibil pe pagina web a Agenţiei Naţionale a Medicamentului şi a Dispozitivelor Medicale din România (www.anm.ro), la secţiunea Raportează o reacţie adversă.
Agenţia Naţională a Medicamentului şi a Dispozitivelor Medicale din România
Str. Aviator Sănătescu nr. 48, sector 1
București 011478-RO
Tel: +4 021 317 11 10
Fax: +4 021 316 34 97
e-mail: adr@anm.ro
Raportare online la adresa: https://adr.anm.ro/
www.anm.ro
Totodată, reacțiile adverse suspectate se pot raporta și către reprezentanța locală a deținătorului autorizației de punere pe piață (DAPP), Teva Pharmaceuticals S.R.L., la următoarele date de contact:
Teva Pharmaceuticals S.R.L.
Calea 13 Septembrie nr. 90, etaj 9, Sector 5, București, România.
Tel: +4021.230.65.24
Fax: +4021. 230.65.23
E-mail: safety.romania@teva-romania.ro
Alina Gabriela Nuta
Departamentul de Farmacovigilență Teva Pharmaceuticals S.R.L.